

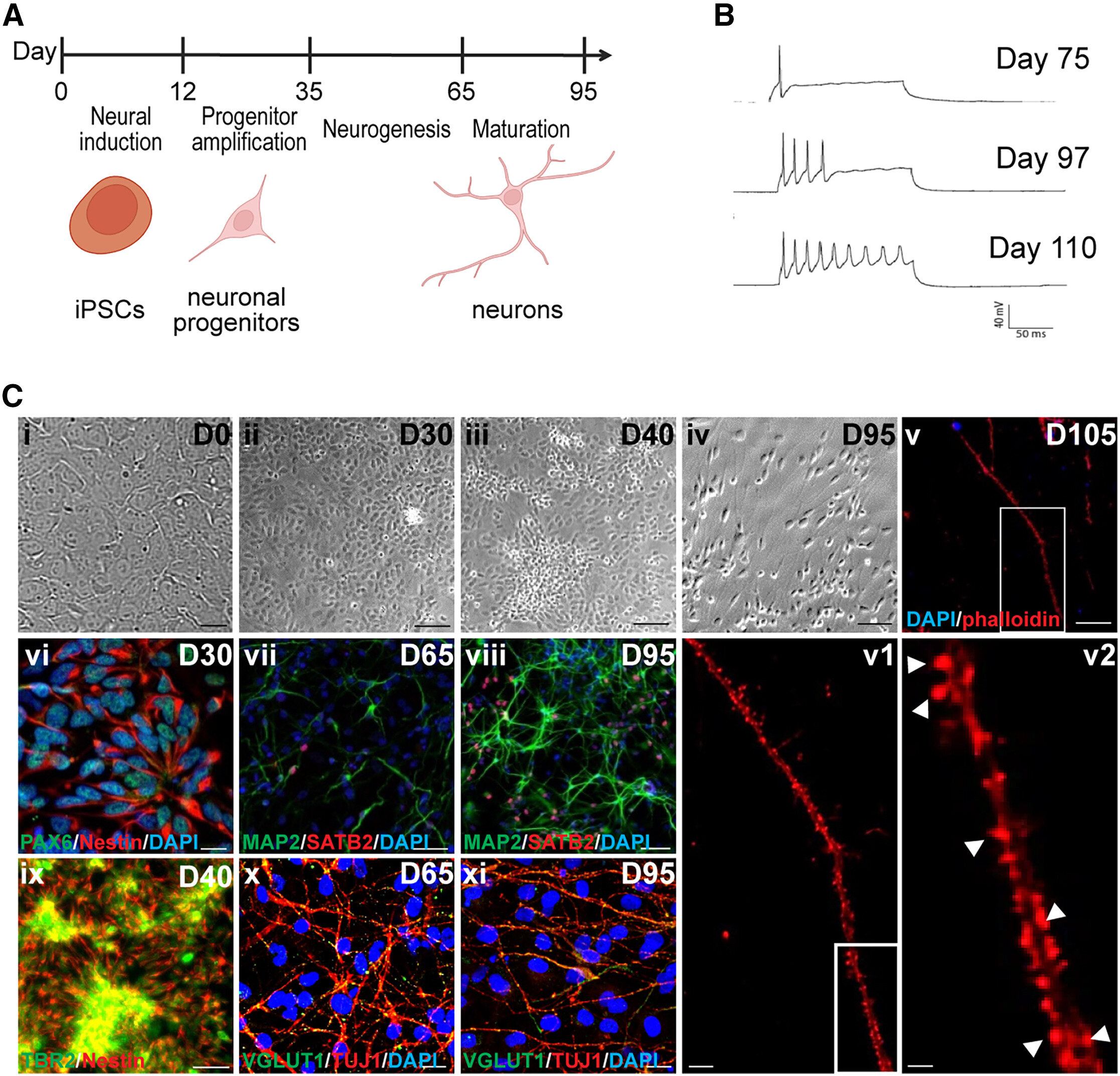
Differentiation of human iPSCs into cortical pyramidal neurons via dual SMAD inhibition. Credit: Stem Cell Reports (2024). DOI: 10.1016/j.stemcr.2024.08.010
Genetic prion disease typically presents with cognitive difficulties, poor muscle control, and sudden jerking movements of muscle groups and/or entire limbs. The three main phenotypes of genetic prion disease are genetic Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), and Gerstmann-Sträussler-Scheinker syndrome (GSS). The most common cause of inherited prion diseases is the E200K mutation of the prion protein (PrP). This mutation is often thought to cause disease by making PrP more likely to misfold into a pathogenic form (PrPSc).
However, new research from the Chobanian & Avedisian School of Medicine at Boston University and Boston Medical Center (BMC) has found that the architecture of neuron-to-neuron contact sites, called synapses, is altered in neurons. expressing this mutant PrP in the absence of PrPSc. This suggests that loss or alteration of PrP function may contribute to the disease phenotype.
The study is published in the journal Stem Cell Reports.
“Our results suggest that there may be detectable abnormalities in neurons long before the main symptoms of inherited prion diseases appear,” explained co-corresponding author David A. Harris, MD, Ph.D. , Edgar Minas Housepian Professor and chair of the school’s Department of Biochemistry and Cell Biology.
Harris and colleagues have built a vast library of induced pluripotent stem cells (iPSCs) (blood cells that have been reprogrammed into an embryonic-like pluripotent state that allows the development of an unlimited source of any human cell type needed for any purpose. therapeutics). from a family harboring this mutation, and differentiated them into neurons.
They then compared the mutations-carrying neurons to non-carrier neurons. They also used CRISPR/Cas9 technology to correct the mutation in two lines, so they could make these comparisons between neurons with identical genetic backgrounds apart from the mutation of interest.
According to the researchers, the use of iPSC technology represents a further step towards personalized medicine. “Our study uses the largest collection of iPSCs from a family harboring an inherited prion disease to our knowledge. iPSC-derived neurons may provide important mechanistic insights into the pathogenesis of genetic prion diseases and may offer a powerful platform for testing candidate therapies,” said co-corresponding author Gustavo Mostoslavsky, MD, Ph.D., professor of medicine and microbiology at the school and co-director of the BU and BMC Center for. Regenerative Medicine.
Researchers believe this study advances the understanding of this rare but devastating group of neurodegenerative disorders that damage the connections between nerve cells in the brain, and provides a guide to treatments that may be most effective in combating the symptoms of these disorders. .
“Similar therapeutic approaches could also be applied to Alzheimer’s disease and other neurodegenerative diseases, some cases of which are hereditary,” Harris said.
More information:
Aldana D. Gojanovich et al, Abnormal synaptic architecture in iPSC-derived neurons from a multigenerational family with genetic Creutzfeldt-Jakob disease, Stem Cell Reports (2024). DOI: 10.1016/j.stemcr.2024.08.010
Provided by Boston University School of Medicine
Quote: Treatments that maintain synapse health may help prevent and alleviate symptoms of prion disease (September 27, 2024) retrieved September 27, 2024 from
This document is subject to copyright. Except for fair use for private study or research purposes, no part may be reproduced without written permission. The content is provided for informational purposes only.

{kind=link}