

Credit: Stem cell (2023). DOI: 10.1016/j.stem.2023.10.007
Researchers from several institutions in China have found a way to use gene editing to reactivate dormant fetal oxygen transport proteins in adult blood cells to potentially reverse a wide range of blood disorders.
In a paper titled “HBG promoter base editing induces potent fetal hemoglobin expression without detectable off-target mutations in human HSCs,” published in Stem cellthe team compares gene editing techniques while formulating a method that could have important clinical applications.
Fetal gammaglobin (γ) is normally replaced by adult hemoglobin (β) during development. In a strange quirk of evolution, only humans and a few types of monkeys are known to switch from expressing γ to β genes.
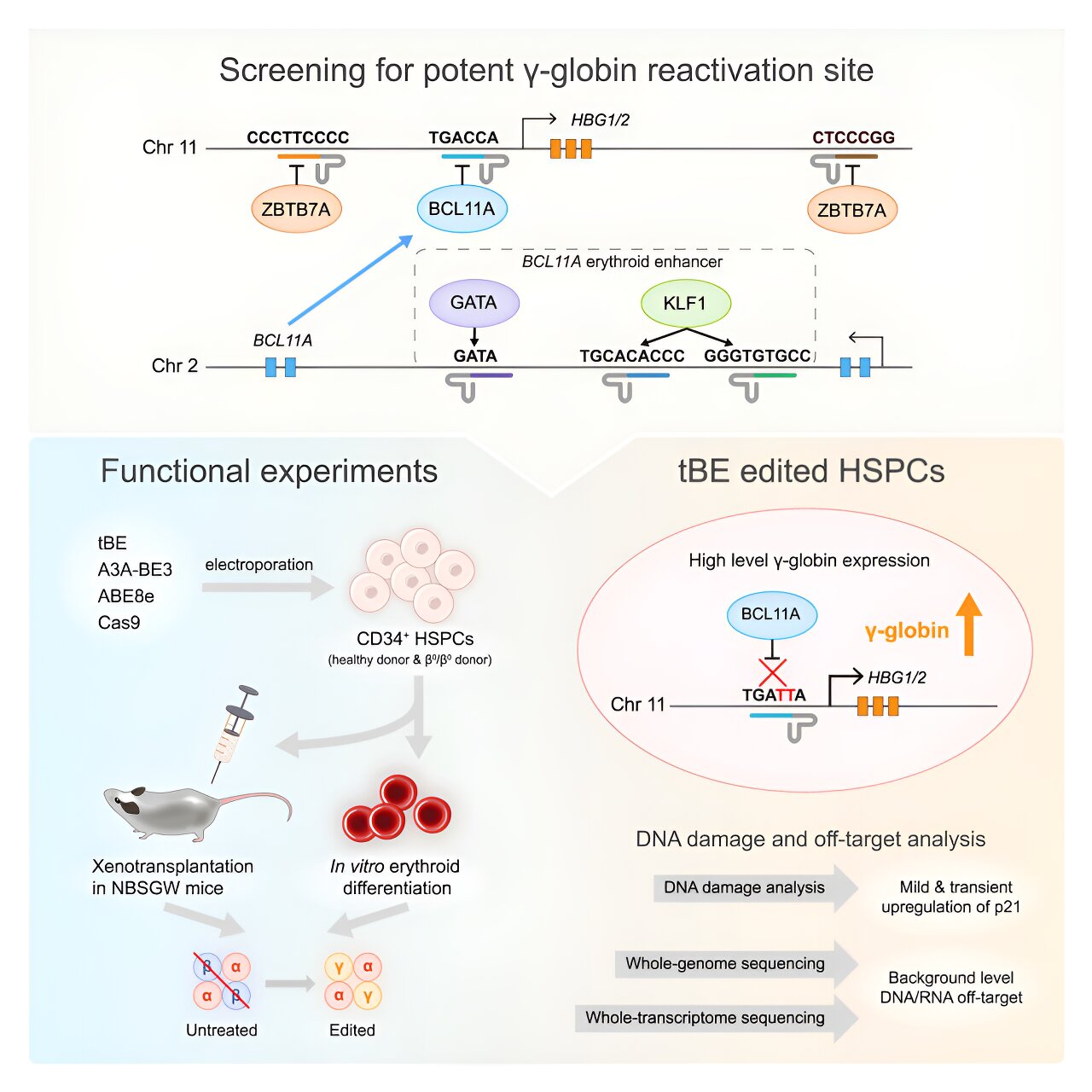
Genes producing fetal hemoglobin become silent and dormant after genetic change by repressors such as BCL11A and ZBTB7A, whose binding motifs have been identified as targets for reactivation.
β-Hemoglobinopathies, including β-thalassemia and sickle cell disease, result from mutations in the HBB gene, leading to impaired β-globin production and leading to anemia, impaired oxygen delivery to tissues and possible multi-organ tissue damage.
Researchers have experimentally discovered that reactivating γ-globin expression could become a universal therapeutic strategy for these conditions.
Six regulatory motifs (BCL11A enhancers and HBG1/2 promoter regions) were targeted using a recently developed cytosine base editor: transformer base editor (tBE). The team compared tBE to other base editors and the Cas9 nuclease to check its effectiveness and off-target effects.
In the study, tBE showed comparable or better editing efficiency than other editors for the targeted patterns. Comprehensive analysis revealed no detectable off-target mutations in tBE-edited cells, indicating the potential of tBE as a safer and more potent treatment strategy for β-hemoglobinopathies.
Experiments with patient-derived cells demonstrated that disruption of BCL11A binding sites within HBG1/2 promoters led to the highest levels of γ-globin expression. Xenotransplantation in mice showed persistent editing of HSCs and their progeny, thereby maintaining engraftment potential and differentiation capacity.
The increased expression of γ-globin observed due to tBE-mediated editing signifies a promising therapeutic avenue for β-hemoglobinopathies.
Although the study focused on editing methods and not direct clinical results, the substantial improvement in γ-globin expression levels strongly suggests potential clinical benefits, including alleviation of symptoms and better disease management for people affected by β-hemoglobinopathies.
More information:
Wenyan Han et al, HBG promoter base editing induces potent fetal hemoglobin expression without detectable off-target mutations in human HSCs, Stem cell (2023). DOI: 10.1016/j.stem.2023.10.007
© 2023 Science X Network
Quote: Reactivation of inhibited fetal hemoglobin genes could fight sickle cell diseases (November 24, 2023) retrieved November 25, 2023 from
This document is subject to copyright. Apart from fair use for private study or research purposes, no part may be reproduced without written permission. The content is provided for information only.

{kind=link}