

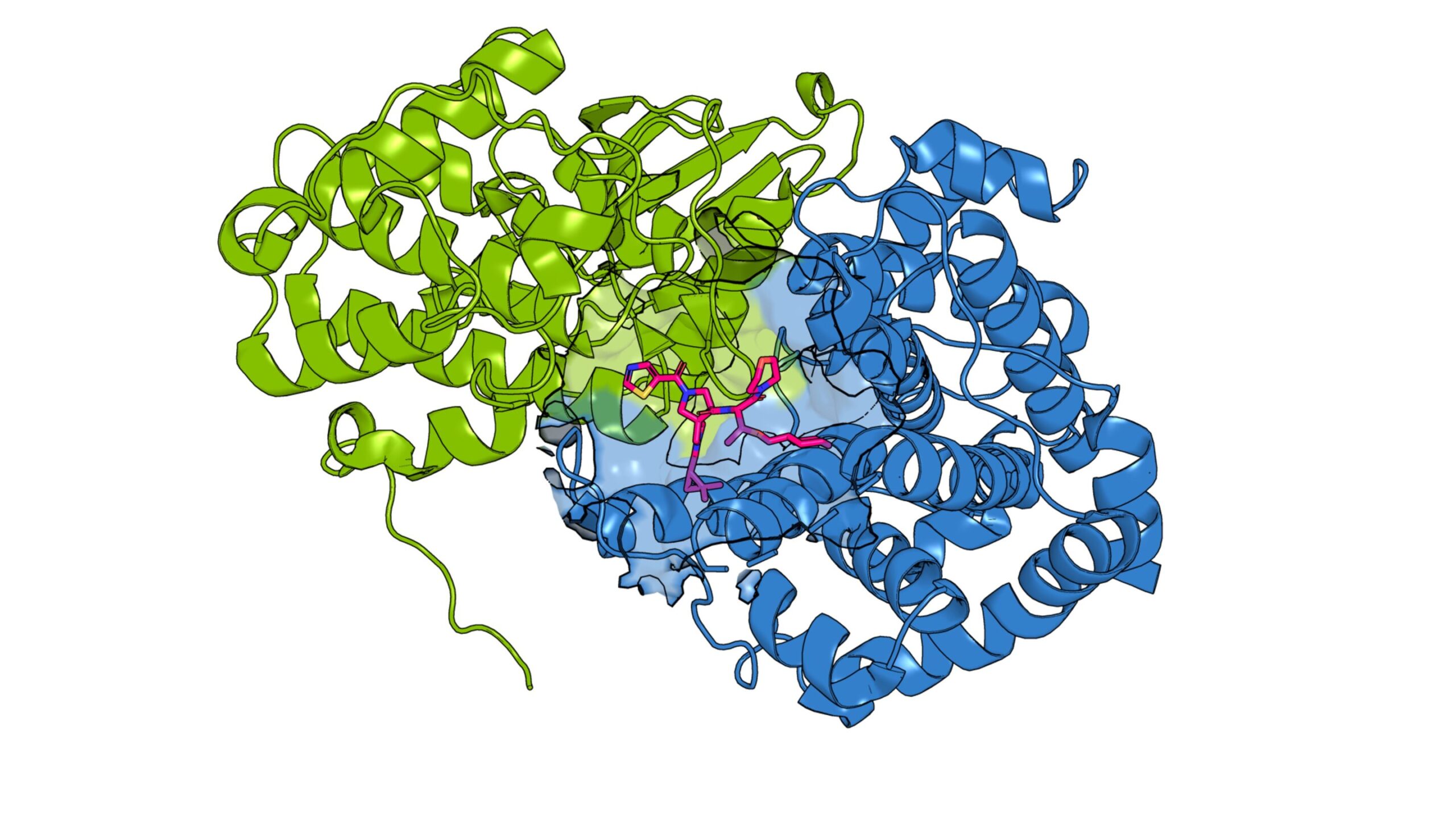
A drug candidate (pink) was found to bind to the pocket between the cancer-related protein CCNE1 (green) and its partner CDK2 (blue) using the novel paralog skipping approach. Credit: Scripps Research
Some proteins in the human body are easy to block with medication; they have an obvious place in their structure where a drug can fit, like a key in a lock. But other proteins are more difficult to target because they lack clear drug-binding sites.
To design a drug that blocks a cancer-linked protein, Scripps Research scientists took inspiration from the protein’s paralog, or “twin.” Using innovative chemical biology methods, the scientists identified a drug site on the paralogue, then used this knowledge to characterize drugs that bound to a similar, but harder to detect, site on its twin. Ultimately, they found drugs that bound only to the protein of interest and not to its very similar sister.
Their approach, described in Nature Chemical Biology on September 18, 2024, and dubbed “paralog jumping,” could uncover new binding sites for drugs and inform drug development more broadly, since nearly half of the proteins in human cells, many involved in cancer and autoimmune diseases have such paralogues.
“This method can be generally useful in cases where you have paralogues and you are trying to find a new drug for one of them,” says lead author Benjamin Cravatt, Ph.D., the Norton B. Gilula Chair in Biology and Chemistry. at Scripps Research. “Being able to target one paralog over another is an important goal in drug development, because two paralogs often have different functions.”
Many genes have duplicated themselves during evolution, resulting in multiple copies in the human genome. In some cases, the copies evolved in slightly different sequences from each other, turning their corresponding proteins into paralogs. These protein paralogs remain very similar in structure and often have redundant or overlapping functions within cells.
In recent years, Cravatt’s research team has formulated an approach to developing drugs that bind to the amino acid cysteine, a protein building block with unique and highly reactive chemical properties. The scientists’ method takes advantage of cysteines as the optimal site for drugs to permanently attach to a protein, often deactivating it. However, not all proteins contain accessible cysteines. In the case of paralogous pairs, one protein may contain a druggable cysteine while the other does not.
“We started with the idea that if you know how to process a protein, you can figure out how to process its paralogue in the same way,” says Yuanjin Zhang, a graduate student at Scripps Research and first author of the new paper.
As a test case, the team addressed the paralog pair known as CCNE1 and CCNE2. Both proteins have been found to be overactive in breast, ovarian and lung cancer. However, scientists suspected that the two proteins played slightly different roles. The team postulated that deactivating a single protein could make treatment of certain cancers more effective.
However, it has been difficult to design drugs targeting CCNE1 and CCNE2 proteins to test this hypothesis. Cravatt, Zhang and their colleagues knew that CCNE2 contained a druggable cysteine, while CCNE1 did not. If they could identify drugs that bound to the same location on CCNE1, even in the absence of cysteine, they suspected the protein would shut down.
The scientists first introduced a cysteine into CCNE1, mimicking the drug binding point they had identified in CCNE2. They then exploited this neo-cysteine to identify drugs that bind to CCNE1. Next, they screened a library of other chemical compounds to determine their ability to compete with this drug by binding to CCNE1. The team reasoned that some of the compounds competing for the same spot would bind in a way that was not dependent on cysteine.
Indeed, Cravatt, Zhang and their colleagues discovered several compounds that could bind to the same site on CCNE1 even when the cysteine was removed again. Some compounds did not bind to CCNE2. Some also had opposite functions, stabilizing the molecule so that it was more active than usual, rather than inactivating it. Structural studies revealed that CCNE1 compounds bind to a cryptic pocket previously not known to be druggable.
The team says the approach highlights the importance of drug testing in diverse and creative ways.
“If we had just looked for compounds with a particular function, we wouldn’t have identified all these different functional molecules, and if we had just looked at the structure of CCNE1, we wouldn’t have found this binding pocket at all ” said Zhang.
Further research is needed to discover whether the new compounds have potential utility in treating cancer or other diseases in which CCNE1 plays a role. Next, the scientists plan to apply their paralog-skipping method to other protein pairs important for tumorigenesis.
In addition to Cravatt and Zhang, the study’s authors include Zhonglin Liu, Sang Joon Won, Divya Bezwada and Bruno Melillo of Scripps; and Marsha Hirschi, Oleg Brodsky, Eric Johnson, Asako Nagata, Matthew D. Petroski, Jaimeen D. Majmudar, Sherry Niessen, Todd VanArsdale, Adam M. Gilbert, Matthew M. Hayward, Al E. Stewart, and Andrew R. Nager of Pfizer , Inc.
More information:
Yuanjin Zhang et al, An allosteric site of cyclin E-CDK2 mapped by paralogous jumps with covalent probes, Nature Chemical Biology (2024). DOI: 10.1038/s41589-024-01738-7
Provided by the Scripps Research Institute
Quote: Seeing double: designing drugs targeting “twin” cancer proteins (October 1, 2024) retrieved October 1, 2024 from
This document is subject to copyright. Except for fair use for private study or research purposes, no part may be reproduced without written permission. The content is provided for informational purposes only.

{kind=link}